Publié le
Lecture 12 mins
Hyperlaxité de l’enfant et de l’adolescent
Bertrand CHEVALLIER, Marlène JOURNEAUX, Centre de référence du Syndrome de Marfan, CHU Bichat (APHP) et Service de pédiatrie, CHU Ambroise-Paré (APHP), Université Paris Saclay
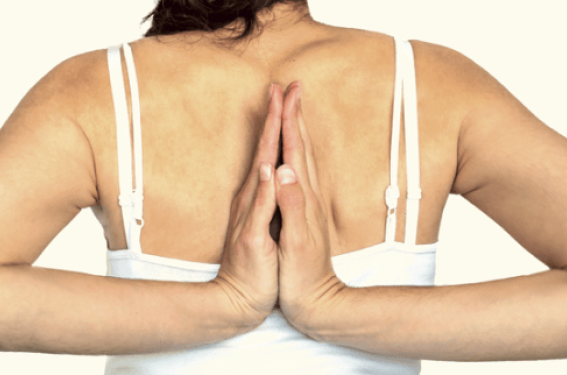
Hyperlaxité et hypermobilité sont fréquentes chez l’enfant, en particulier chez les plus jeunes entre 1 et 4 ans. Quelle démarche diagnostic suivre ? Quand rechercher une cause et notamment quand évoquer un syndrome d’Ehlers-Danlos ?
Les réponses de Bertrand Chevallier et de Marlène Journeaux (Boulogne-Billancourt).
Les termes hypermobilité, hyperlaxité, sont utilisés indifféremment par certains auteurs. L’hypermobilité traduit communément l’augmentation d’une amplitude articulaire physiologique, tandis que l’hyperlaxité traduit un mouvement excessif dans un plan de mobilité articulaire anormal. Une...
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :